
Multifocal Atrophies & Subsystem Disruption in Alzheimer’s Disease
Frontotemporal Atrophy and Case FG
There is a subgroup of AD cases with frontal lobe glucose hypometabolism and a high degree of behavioral deficits typical of frontal lobe damage. Grady et al. (1990) observed more inappropriate behaviors in cases with greatest prefrontal glucose hypometabolism. Mann et al. (1992b) reported that two groups with equivalent parietal glucose hypometabolism and impaired neuropsychological measures of parietal function could be distinguished in terms of frontal lobe glucose metabolism, measures of executive functions, and rate of disease progression. Cases with most rapid progression and impaired executive functions had greatest glucose hypometabolism in prefrontal cortex. Finally, Royall et al. (1994) observed early impairment of executive control functions in AD that could be attributed to disrupted frontal lobe functions.
Although large neuron degeneration has been reported in mid-prefrontal cortex, there is a wide range of degeneration, with many cases having a normal density of neurons (Terry et al., 1981). Brun and Englund (1981) report only modest losses of neurons in prefrontal cortex. Furthermore, there is heterogeneity in NFT densities, which are significantly different for early- versus late-onset cases (Hansen et al., 1988)/ In the context of disease heterogeneity, frontal cortex is a region that is differentially impacted by AD and in cases where frontotemporal atrophy is particularly striking and associated with behavioral deficits that suggest a clinicopathological subtype.
Case FG is an example of AD with focal frontotemporal degeneration. FG was an intelligent, right-handed male who was an electrical engineer. At the age of 85, he was driving the family on a long trip, during which he became profoundly paranoid of truck drivers to the point that he would follow them to verify that they were not endangering the family. Upon arrival at their destination, he immediately returned to the car to continue driving to another destination. Within two years he was evaluated neurologically and said to have increased problems with spatial disorientation, recent memory, and forgetfulness. His immediate recall was fairly good, but he had a problem remembering current events. No active hallucinations or delusions were noted at this time. The CT scans showed mild to moderate cortical atrophy and ventricular enlargement, findings that were confirmed one year later, as shown in Figure 2. At 89 FG suffered from mood instability and physical aggression toward his wife. In the past tow years he showed many menacing behaviors such as threatening to hit his wife with a metal cane or kitchen chair. He was over responsive to and agitated by TV violence to the point that the TV was removed from his room. He was frequently concerned about "people being around and coming into the house." On one visit, a psychiatrist observed that FG was alert, semi-cooperative, and well groomed, although he was wearing a leather jacket, shirt, and sweater all buttoned and tucked into his pants. There was mild psychomotor agitation, and he attempted to leave the examination and resisted being redirected. There was some loss of facial expression, but speech was at a normal rate, volume, and tone. Mood was euthymic with an appropriate, bright and stable affect. His thought was severely disorganized, with most responses being incoherent and nonsensical. He had visual and auditory hallucinations. His praxis was poor and he could not name colors and misnamed a pen, but was able to name a watch and knew his name. Neuropsychological tests showed global deterioration in all cognitive domains, including attention, memory, registration, and recall, as well as language, naming, praxis/executive function, and perceptual disturbances. Speech had declined to where complete sentences were not always used, and he frequently spoke with adjectives until his death at age 90.
Postmortem assessment showed profound frontotemporal atrophy (Fig 2B). There were no Lewy bodies, Pick bodies, or ballooned neurons in prefrontal or any other neocortical region assessed, suggesting that this was not a case of frontal lobe dementia. The ApoE genotype was homozygous for the e4 allele and this is an infrequent finding in frontal lobe dementia (Pickering-Brown et al., 1995). There was a high density of SP throughout all neocortical regions, including prefrontal, inferior parietal, and PCC. Throughout neocortex there were a limited number of layer V NFT and in the hippocampal information and entorhinal/perirhinal cortices there was a pattern of NFT "typical" of AD. Heavy Ab42 deposition in all cortical regions, including prominent buildup in prefrontal cortex, underscored the AD pathogenesis of neocortical neurodegeneration.
Tau-ir NFT were particularly high around the fundus of midcingulate cortex (Fig.2, area 24d). This region had a high density of NFT in layer V and numerous neuritic plaques in layers I-III. This region contains the caudal cingulate motor area and it is interesting in this context because FG suffered from dysexecutive syndrome and motor system disinhibition. Also of interest in terms of the cingulate gyrus is that area 23a had equal densities of NFT in layers V and II-IIIab, and area 23 had the highest density of neuropil threads in neocortex (Fig. 2).
There is a subgroup of AD cases with frontal lobe glucose hypometabolism and a high degree of behavioral deficits typical of frontal lobe damage. Grady et al. (1990) observed more inappropriate behaviors in cases with greatest prefrontal glucose hypometabolism. Mann et al. (1992b) reported that two groups with equivalent parietal glucose hypometabolism and impaired neuropsychological measures of parietal function could be distinguished in terms of frontal lobe glucose metabolism, measures of executive functions, and rate of disease progression. Cases with most rapid progression and impaired executive functions had greatest glucose hypometabolism in prefrontal cortex. Finally, Royall et al. (1994) observed early impairment of executive control functions in AD that could be attributed to disrupted frontal lobe functions.
Although large neuron degeneration has been reported in mid-prefrontal cortex, there is a wide range of degeneration, with many cases having a normal density of neurons (Terry et al., 1981). Brun and Englund (1981) report only modest losses of neurons in prefrontal cortex. Furthermore, there is heterogeneity in NFT densities, which are significantly different for early- versus late-onset cases (Hansen et al., 1988)/ In the context of disease heterogeneity, frontal cortex is a region that is differentially impacted by AD and in cases where frontotemporal atrophy is particularly striking and associated with behavioral deficits that suggest a clinicopathological subtype.
Case FG is an example of AD with focal frontotemporal degeneration. FG was an intelligent, right-handed male who was an electrical engineer. At the age of 85, he was driving the family on a long trip, during which he became profoundly paranoid of truck drivers to the point that he would follow them to verify that they were not endangering the family. Upon arrival at their destination, he immediately returned to the car to continue driving to another destination. Within two years he was evaluated neurologically and said to have increased problems with spatial disorientation, recent memory, and forgetfulness. His immediate recall was fairly good, but he had a problem remembering current events. No active hallucinations or delusions were noted at this time. The CT scans showed mild to moderate cortical atrophy and ventricular enlargement, findings that were confirmed one year later, as shown in Figure 2. At 89 FG suffered from mood instability and physical aggression toward his wife. In the past tow years he showed many menacing behaviors such as threatening to hit his wife with a metal cane or kitchen chair. He was over responsive to and agitated by TV violence to the point that the TV was removed from his room. He was frequently concerned about "people being around and coming into the house." On one visit, a psychiatrist observed that FG was alert, semi-cooperative, and well groomed, although he was wearing a leather jacket, shirt, and sweater all buttoned and tucked into his pants. There was mild psychomotor agitation, and he attempted to leave the examination and resisted being redirected. There was some loss of facial expression, but speech was at a normal rate, volume, and tone. Mood was euthymic with an appropriate, bright and stable affect. His thought was severely disorganized, with most responses being incoherent and nonsensical. He had visual and auditory hallucinations. His praxis was poor and he could not name colors and misnamed a pen, but was able to name a watch and knew his name. Neuropsychological tests showed global deterioration in all cognitive domains, including attention, memory, registration, and recall, as well as language, naming, praxis/executive function, and perceptual disturbances. Speech had declined to where complete sentences were not always used, and he frequently spoke with adjectives until his death at age 90.
Postmortem assessment showed profound frontotemporal atrophy (Fig 2B). There were no Lewy bodies, Pick bodies, or ballooned neurons in prefrontal or any other neocortical region assessed, suggesting that this was not a case of frontal lobe dementia. The ApoE genotype was homozygous for the e4 allele and this is an infrequent finding in frontal lobe dementia (Pickering-Brown et al., 1995). There was a high density of SP throughout all neocortical regions, including prefrontal, inferior parietal, and PCC. Throughout neocortex there were a limited number of layer V NFT and in the hippocampal information and entorhinal/perirhinal cortices there was a pattern of NFT "typical" of AD. Heavy Ab42 deposition in all cortical regions, including prominent buildup in prefrontal cortex, underscored the AD pathogenesis of neocortical neurodegeneration.
Tau-ir NFT were particularly high around the fundus of midcingulate cortex (Fig.2, area 24d). This region had a high density of NFT in layer V and numerous neuritic plaques in layers I-III. This region contains the caudal cingulate motor area and it is interesting in this context because FG suffered from dysexecutive syndrome and motor system disinhibition. Also of interest in terms of the cingulate gyrus is that area 23a had equal densities of NFT in layers V and II-IIIab, and area 23 had the highest density of neuropil threads in neocortex (Fig. 2).
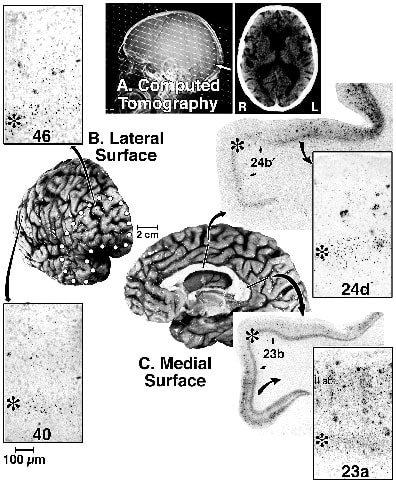
Figure 2. Case FG is an example of AD with focal frontotemporal atrophy. (A) Computed tomography two years before death shows mild cerebral atrophy and ventricular swelling. Postmortem assessment demonstrated clear frontotemporal atrophy (B, outlined with dots) and moderate dilation of sulci on the superior and medial surface (C). Insets are tau-immunoreactive NFT and neuritic plaques in areas 46, 40, 24d, and 23a. Greatest NFT numbers were in layer V (asterisks). Area 23a had the highest density of neuropil threads of any neocortical area and equal density of NFT in layers IIIab and Va. The highest level of neurofibrillary degeneration so far observed in this case is in the fundus of the midcingulate sulcus, suggesting involvement of the caudal cingulate motor area.
The density of thioflavine S-stained, mature, and burned-out SP was about the same in all neocortices. Immunohistochemistry for antibodies selective for Ab40 and Ab42 showed that the clumps of thioflavine S-stained, mature, and burned-out SP were similar to those immunoreactive for Ab40. The very substantial neurodegeneration in this case was in layers II and III as shown for area 46 in Figure 3 and there was a striking match between this cell loss and uniform deposition of Ab42 in layer III. Notice in area 46 that the highest levels of Ab42 were in layers II-IIIab, as indicated with the bracket and these layers are the site of greatest neurodegeneration in area 46.
The density of thioflavine S-stained, mature, and burned-out SP was about the same in all neocortices. Immunohistochemistry for antibodies selective for Ab40 and Ab42 showed that the clumps of thioflavine S-stained, mature, and burned-out SP were similar to those immunoreactive for Ab40. The very substantial neurodegeneration in this case was in layers II and III as shown for area 46 in Figure 3 and there was a striking match between this cell loss and uniform deposition of Ab42 in layer III. Notice in area 46 that the highest levels of Ab42 were in layers II-IIIab, as indicated with the bracket and these layers are the site of greatest neurodegeneration in area 46.
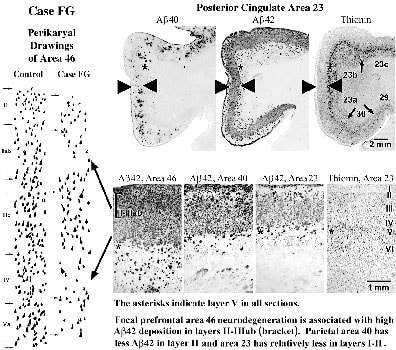
Figure 3. Drawings of neuronal perikarya through prefrontal area 46 in a control case and case FG. This area is atrophied and shows substantial neurodegeneration, particularly in layers IIIab, IV, and V. Ab40 and Ab42 immunohistochemistry in PCC is shown and for prefrontal area 46 and parietal area 40. Ab40-ir mature and burned out plaques form clumps in layer III of all areas and parallels the appearance of thioflavine S-stained SP. Ab42 has a uniform distribution in all areas and is associated mainly with diffuse and immature plaques in layer III and mature plaques in layers I-II and V-VI. Although many neurons are lost throughout cortex, most dense deposits of Ab42 are in layers II-IIIab of area 46, where gross focal atrophy and neurodengeneration occur as emphasized with the arrows. There is a clear association in PCC of neuron degeneration in layers II-III and maximal deposits of Ab42 and this is emphasized with large white arrows in layers II-III.
Most neuron losses in areas 46 and 23 were in superficial II-IIIab and deep layers V and VI. In area 40, neurodegeneration was significant in layers IV and V, however, layers II-III had much less degeneration, and there was no overt cortical shrinkage. The striking association between focal prefrontal atrophy, the highest levels of Ab42, and a match between highest Ab42 and neurodegeneration in layers II-IIIab suggests that Ab42 neurotoxicity may be a key factor in multifocal cortical atrophies. Not all cell loss, however, was related to Ab42 early on. In area 23, layers II-IIIab also had a high number of NFT. Layer IV had severe neuron losses in all areas, but only moderate Ab42 deposition, while neurons in layer IIIc were relatively free of NFT and degeneration. There were neuron losses in deeper layers with only scattered Ab42-ir SP, but this could have been due to neurofibrillary changes or the actions of Ab42 on the apical dendrites of pyramidals neurons.
Overall, it is unlikely that a single mechanism of neurodegeneration was responsible for these complex patterns of neurodegeneration and disease markers. The severe neurodegeneration in prefrontotemporal and cingulate areas likely impaired the regulation of executive motor functions. Although the extensive degeneration in these regions is only partially accounted for by NFT density, there are some close matches between the deposition of Ab42 and neurodegeneration that may lead to an understanding of the mechanisms of focal cortical neurodegeneration and subtypes of the disease.
Most neuron losses in areas 46 and 23 were in superficial II-IIIab and deep layers V and VI. In area 40, neurodegeneration was significant in layers IV and V, however, layers II-III had much less degeneration, and there was no overt cortical shrinkage. The striking association between focal prefrontal atrophy, the highest levels of Ab42, and a match between highest Ab42 and neurodegeneration in layers II-IIIab suggests that Ab42 neurotoxicity may be a key factor in multifocal cortical atrophies. Not all cell loss, however, was related to Ab42 early on. In area 23, layers II-IIIab also had a high number of NFT. Layer IV had severe neuron losses in all areas, but only moderate Ab42 deposition, while neurons in layer IIIc were relatively free of NFT and degeneration. There were neuron losses in deeper layers with only scattered Ab42-ir SP, but this could have been due to neurofibrillary changes or the actions of Ab42 on the apical dendrites of pyramidals neurons.
Overall, it is unlikely that a single mechanism of neurodegeneration was responsible for these complex patterns of neurodegeneration and disease markers. The severe neurodegeneration in prefrontotemporal and cingulate areas likely impaired the regulation of executive motor functions. Although the extensive degeneration in these regions is only partially accounted for by NFT density, there are some close matches between the deposition of Ab42 and neurodegeneration that may lead to an understanding of the mechanisms of focal cortical neurodegeneration and subtypes of the disease.
Parietotemporal Atrophy and Case WJ
The "typical" case of AD is characterized by bilateral hypometabolism of the medial temporal region, and posterior temporal and parietal cortices, with associated deficits in learning, memory, language abilities, and visuospatial skills, respectively. Some patients, however, may have deficits limited to word-finding and other language abilities and show relatively focal hypometabolism of the left temporal lobe, or to visuospatial abilities, with hypometabolism limited to parietal cortex. Case WJ had early language deficits and severe left temporal pole atrophy that later advanced to a visuospatial deficit and a swath of parietotemporal atrophy at postmortem. Although this case had more extensive cortical involvement than case FG, it provides insight into neuronal substrates of cortical function.
The first sign of a cognitive problem in case WJ was impairment in word finding. Magnetic resonance imaging subsequently showed evidence of profound temporal atrophy in the left hemisphere, and the most severe atrophy at postmortem was in the same region (Fig. 4). Because the language impairment began years before the memory and visuospatial impairments, the first sites of disrupted neuronal function may have been in superior temporal and temporal pole cortices rather than in hippocampal, entorhinal, and parietal cortices. Case WJ was a right-handed male with a Ph.D. in biochemistry. He was very athletic, enjoyed frequent outdoor trips, and did not use alcohol or smoke. When he was about 67 his wife noted that he had word-finding problems, substitution of the wrong words, and spelling and pronunciation difficulties. Her experiences in special education and her husband’s high level of education made these changes more easily identified than might have been the case in other families. It is also interesting that his mother had possible AD, and WJ’s wife reported that his dementia did not appear to follow a similar course to his mother’s. Two years later he was taking a cross country camping trip alone and got lost because he took wrong turns and could not read maps correctly. By age 74 he was speaking with pronouns only rather than nouns and he could not write. He would get lost going to a local gasoline station. Neurological assessment at that time was unremarkable beyond the dementing illness, and CT showed mild cortical atrophy in addition to severe atrophy in the left temporal pole. Within one year MR studies confirmed the earlier CT observations and it was noted that there were bilateral areas of white matter high signal intensities, particularly underlying parietal cortex. Profound left-hemisphere atrophy and bilateral leukoaraiosis can be seen in Figure 4. A 7-month treatment with Cognex did not alter the progression of WJ’s language and visuospatial deficits. There was no evidence of behavioral impairments characteristic of case FG, nor oculomotor deficits encountered in posterior cortical atrophy in AD, as discussed below. To the time of his death at age 77, WJ had a relatively restricted cognitive deficit.
The first sign of a cognitive problem in case WJ was impairment in word finding. Magnetic resonance imaging subsequently showed evidence of profound temporal atrophy in the left hemisphere, and the most severe atrophy at postmortem was in the same region (Fig. 4). Because the language impairment began years before the memory and visuospatial impairments, the first sites of disrupted neuronal function may have been in superior temporal and temporal pole cortices rather than in hippocampal, entorhinal, and parietal cortices. Case WJ was a right-handed male with a Ph.D. in biochemistry. He was very athletic, enjoyed frequent outdoor trips, and did not use alcohol or smoke. When he was about 67 his wife noted that he had word-finding problems, substitution of the wrong words, and spelling and pronunciation difficulties. Her experiences in special education and her husband’s high level of education made these changes more easily identified than might have been the case in other families. It is also interesting that his mother had possible AD, and WJ’s wife reported that his dementia did not appear to follow a similar course to his mother’s. Two years later he was taking a cross country camping trip alone and got lost because he took wrong turns and could not read maps correctly. By age 74 he was speaking with pronouns only rather than nouns and he could not write. He would get lost going to a local gasoline station. Neurological assessment at that time was unremarkable beyond the dementing illness, and CT showed mild cortical atrophy in addition to severe atrophy in the left temporal pole. Within one year MR studies confirmed the earlier CT observations and it was noted that there were bilateral areas of white matter high signal intensities, particularly underlying parietal cortex. Profound left-hemisphere atrophy and bilateral leukoaraiosis can be seen in Figure 4. A 7-month treatment with Cognex did not alter the progression of WJ’s language and visuospatial deficits. There was no evidence of behavioral impairments characteristic of case FG, nor oculomotor deficits encountered in posterior cortical atrophy in AD, as discussed below. To the time of his death at age 77, WJ had a relatively restricted cognitive deficit.
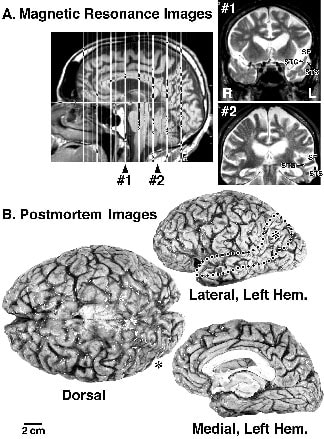
Figure 4. Case WJ is an example of focal parietotemporal atrophy in AD. (A) Magnetic resonance images taken 3 years before death. Section 1 shows the severe left-temporal pole atrophy, and section 2 shows the severe superior temporal gyrus atrophy. (B) The postmortem images confirm the region of focal atrophy (outlined with circles). The asterisk on the left lateral surface is on posterior parietal cortex. The dorsal view can be used to compare the left and right hemispheres. The medial hemisphere has significant atrophy as seen by widening of vertically oriented sulci along the superior surface, including the paracentral and parieto-occipital sulci and marginal ramus of the cingulate sulcus. The left medial surface was reoriented for ease of comparison of regions with atrophy.
At postmortem the brain weighed 1320 gm and had severe left-hemisphere atrophy, particularly in parietotemporal cortex (Fig. 4). There was a relative preservation of most of prefrontal cortex bilaterally except for area 10 of the frontal pole. Atrophy in the temporal pole was profound and localized to the left hemisphere (asterisk in Fig. 4). The medial surface had a moderate degree of atrophy, with widening of a number of the vertically oriented sulci including the marginal ramus of the cingulate sulcus. Histological analyses showed that the high-signal intensity in the white matter were areas of watery astrocytosis associated with severe axon degeneration. The density of thioflavine S-stained SP was typical for advanced stages of AD and relatively uniform throughout neocortex. Subcortical structures appeared normal and the locus coeruleus and raphe nuclei had normal densities of neurons, although some reduction in pigmentation, and occasional NFT. Neocortical neurodegeneration was severe throughout the left hemisphere, including areas 10, 40, and 23. There was almost no laminar architecture and neurons mainly in layer IIIc were preserved, and provided landmarks for assessing neuron densities.
This case documents and early language deficit in AD that is associated with profound and focal atrophy in left superior temporal and temporal pole cortices. Tyrrell et al. (1990) report significant glucose hypometabolism in left anterior temporal lobe associated with progressive aphasia in cases with focal cortical atrophy. Their study supports the view that rostral, left-temporal atrophy contributed to the language deficit in case WJ. The disease in WJ appears to have spread from this region into parietal cortex within two years of the onset of language impairment, contributing the visuospatial deficit and producing an enlarged area of atrophy at lateral neocortical areas, and raises the question regarding the extent to which focal lobar damage and disruption of functional subsystems in AD represent etiologically unique subtypes.
At postmortem the brain weighed 1320 gm and had severe left-hemisphere atrophy, particularly in parietotemporal cortex (Fig. 4). There was a relative preservation of most of prefrontal cortex bilaterally except for area 10 of the frontal pole. Atrophy in the temporal pole was profound and localized to the left hemisphere (asterisk in Fig. 4). The medial surface had a moderate degree of atrophy, with widening of a number of the vertically oriented sulci including the marginal ramus of the cingulate sulcus. Histological analyses showed that the high-signal intensity in the white matter were areas of watery astrocytosis associated with severe axon degeneration. The density of thioflavine S-stained SP was typical for advanced stages of AD and relatively uniform throughout neocortex. Subcortical structures appeared normal and the locus coeruleus and raphe nuclei had normal densities of neurons, although some reduction in pigmentation, and occasional NFT. Neocortical neurodegeneration was severe throughout the left hemisphere, including areas 10, 40, and 23. There was almost no laminar architecture and neurons mainly in layer IIIc were preserved, and provided landmarks for assessing neuron densities.
This case documents and early language deficit in AD that is associated with profound and focal atrophy in left superior temporal and temporal pole cortices. Tyrrell et al. (1990) report significant glucose hypometabolism in left anterior temporal lobe associated with progressive aphasia in cases with focal cortical atrophy. Their study supports the view that rostral, left-temporal atrophy contributed to the language deficit in case WJ. The disease in WJ appears to have spread from this region into parietal cortex within two years of the onset of language impairment, contributing the visuospatial deficit and producing an enlarged area of atrophy at lateral neocortical areas, and raises the question regarding the extent to which focal lobar damage and disruption of functional subsystems in AD represent etiologically unique subtypes.
Posterior Cortical Atrophy
Although AD patients who first experience visuospatial deficits tend to have an earlier onset of the disease and appear to be less frequent than the typical pattern of early memory impairment (Albert et al., 1990), they are of particular interest because early visuospatial deficits are often associated with posterior cortical atrophy in AD (Benson et al., 1988). This contrasts with the common pattern of AD pathology that tends to have less severely affected visual occipital areas in comparison to frontal, parietal, and temporal cortices (Arnold et al., 1991). In addition, the significant involvement of PCC in these cases (Hof et al., 1993a, 1997; Vogt et al., 1997) raises the possibility that they also represent a unique subtype.
The visual impairment is not diffuse, but rather affects specific aspects of visual function, and it is important to discuss the selective involvement of particular components of the visual circuitry in the neurodegenerative process in AD. Several clinical and neuropathological studies have demonstrated the occurrence of AD cases with atypical neuro-ophthalmological presentation (Cogan, 1985; Morel, 1945; Fletcher, 1994; Benson et al., 1988). Most patients with posterior cortical atrophy initially display a complex visual syndrome characterized by paralysis of visual fixation associated with an optic ataxia and a disturbance of visual attention known as Bálint’s syndrome (Bálint, 1909; Grünthal, 1928; Benson et al., 1988; Fletcher, 1994). Deficits in motion perception and target tracing are usual, although not always consistent (Neary and Snowden, 1987; Fletcher and Sharpe, 1988, Kiyosawa et al., 1989; Kurylo et al., 1994; Butter et al., 1996).
The posterior cortical atrophy in this syndrome is shown in Figure 5A. This distribution of gross atrophy has been demonstrated with CT and MRI, and at autopsy, and it predominates in the parietotemporo-occipital junction and in occipital and posterior cingulate cortices (Nissen et al., 1985; Benson et al., 1988; Berthier et al., 1991; Hof and Bouras, 1991; Hof et al., 1989, 1990a, 1993a; Mendez et al., 1990a,b,c; Graff-Radford et al., 1993; Levine et al., 1993; Levy et al., 1995; Pietrini et al., 1996). The clinical symptomatology of AD patients with posterior cortical atrophy suggests involvement of cortical pathways linking the primary visual regions to the posterior parietal, cingulate, and visual association cortices in the early stages of the dementia (Hof and Bouras, 1991; Hof et al., 1989, 1990a, 1993a, 1997). There is a gradient of NFT densities from area V1 to the visual association regions in the parietal cortex, as well as in the PCC in these cases, all of whom suffered from visuomotor impairment resembling Bálint’s syndrome. The same gradient is apparent with Ab-ir.
In cases with visuomotor impairment, the number of NFT in layers II-III and V-VI is almost twice as high in area V2 as in V1, and further increases in areas V3/V4, 7b/7m, and 23. Also, prefrontal areas 9, 46, and 45 have far fewer NFT than the occipital (except V1), parietal, and posterior cingulate regions, indicating a shift of NFT distribution onto the occipital and parietal cortices. In these cases V1 and V2 contain comparable NFT densities, and in some cases up to three times as high NFT densities are in areas V3/V4, MT, 7b/7m, and 23, where they can be as high as 8.5-fold in layers II-III and 5.4-fold in layers V-VI. Figure 5B shows the severe neurofibrillary changes in areas 29, 30, and 23a of PCC and the relatively smaller involvement of medial parietal area 7. In these cases, the inferior temporal cortex contains higher NFT counts than prefrontal areas, yet the densities in areas TE and TEO are lower than those in the posterior cingulate and inferior parietal cortices, particularly area MT. Finally, SP distribution also has a progressive density increase form area V1 to areas V2, V3/V4, MT, 7b/7m, and 23. Area V2 contains about 30% more SP than area VI. Layers I-III of areas MT and 23 show the highest SP densities.
These patterns of NFT may correspond to functional systems in human occipital cortex (Watson et al., 1993; McCarthy et al., 1995; Sereno et al., 1995; Totell et al., 1995; DeYoe et al., 1996). In particular, the region corresponding to area MT consistently contains more NFT than cortex of the occipital fields. The region reported by Orban et al. (1995) involved in detecting kinetic contours may correspond to the inferotemporal and medial parietal areas 7b/7m that are severely affected in posterior cortical atrophy. The consistent and severe involvement of PCC is particularly relevant in light of recent observations that this area is involved in visuospatial functions. Neurons in monkey area 23 respond to visual stimuli of large textured patterns and the orbital position of the eye (Olson et al., 1993), while lesions in PCC impair spatial memory (Murray et al., 1989). These data suggest that cortical networks linking the parietal, cingulate, and occipital regions are involved in AD and are selectively affected with posterior cortical atrophy (Hof et al., 1993a, 1997).
The visual impairment is not diffuse, but rather affects specific aspects of visual function, and it is important to discuss the selective involvement of particular components of the visual circuitry in the neurodegenerative process in AD. Several clinical and neuropathological studies have demonstrated the occurrence of AD cases with atypical neuro-ophthalmological presentation (Cogan, 1985; Morel, 1945; Fletcher, 1994; Benson et al., 1988). Most patients with posterior cortical atrophy initially display a complex visual syndrome characterized by paralysis of visual fixation associated with an optic ataxia and a disturbance of visual attention known as Bálint’s syndrome (Bálint, 1909; Grünthal, 1928; Benson et al., 1988; Fletcher, 1994). Deficits in motion perception and target tracing are usual, although not always consistent (Neary and Snowden, 1987; Fletcher and Sharpe, 1988, Kiyosawa et al., 1989; Kurylo et al., 1994; Butter et al., 1996).
The posterior cortical atrophy in this syndrome is shown in Figure 5A. This distribution of gross atrophy has been demonstrated with CT and MRI, and at autopsy, and it predominates in the parietotemporo-occipital junction and in occipital and posterior cingulate cortices (Nissen et al., 1985; Benson et al., 1988; Berthier et al., 1991; Hof and Bouras, 1991; Hof et al., 1989, 1990a, 1993a; Mendez et al., 1990a,b,c; Graff-Radford et al., 1993; Levine et al., 1993; Levy et al., 1995; Pietrini et al., 1996). The clinical symptomatology of AD patients with posterior cortical atrophy suggests involvement of cortical pathways linking the primary visual regions to the posterior parietal, cingulate, and visual association cortices in the early stages of the dementia (Hof and Bouras, 1991; Hof et al., 1989, 1990a, 1993a, 1997). There is a gradient of NFT densities from area V1 to the visual association regions in the parietal cortex, as well as in the PCC in these cases, all of whom suffered from visuomotor impairment resembling Bálint’s syndrome. The same gradient is apparent with Ab-ir.
In cases with visuomotor impairment, the number of NFT in layers II-III and V-VI is almost twice as high in area V2 as in V1, and further increases in areas V3/V4, 7b/7m, and 23. Also, prefrontal areas 9, 46, and 45 have far fewer NFT than the occipital (except V1), parietal, and posterior cingulate regions, indicating a shift of NFT distribution onto the occipital and parietal cortices. In these cases V1 and V2 contain comparable NFT densities, and in some cases up to three times as high NFT densities are in areas V3/V4, MT, 7b/7m, and 23, where they can be as high as 8.5-fold in layers II-III and 5.4-fold in layers V-VI. Figure 5B shows the severe neurofibrillary changes in areas 29, 30, and 23a of PCC and the relatively smaller involvement of medial parietal area 7. In these cases, the inferior temporal cortex contains higher NFT counts than prefrontal areas, yet the densities in areas TE and TEO are lower than those in the posterior cingulate and inferior parietal cortices, particularly area MT. Finally, SP distribution also has a progressive density increase form area V1 to areas V2, V3/V4, MT, 7b/7m, and 23. Area V2 contains about 30% more SP than area VI. Layers I-III of areas MT and 23 show the highest SP densities.
These patterns of NFT may correspond to functional systems in human occipital cortex (Watson et al., 1993; McCarthy et al., 1995; Sereno et al., 1995; Totell et al., 1995; DeYoe et al., 1996). In particular, the region corresponding to area MT consistently contains more NFT than cortex of the occipital fields. The region reported by Orban et al. (1995) involved in detecting kinetic contours may correspond to the inferotemporal and medial parietal areas 7b/7m that are severely affected in posterior cortical atrophy. The consistent and severe involvement of PCC is particularly relevant in light of recent observations that this area is involved in visuospatial functions. Neurons in monkey area 23 respond to visual stimuli of large textured patterns and the orbital position of the eye (Olson et al., 1993), while lesions in PCC impair spatial memory (Murray et al., 1989). These data suggest that cortical networks linking the parietal, cingulate, and occipital regions are involved in AD and are selectively affected with posterior cortical atrophy (Hof et al., 1993a, 1997).

Figure 5. Two cases of AD with posterior cortical atrophy. (A) Surface morphology of a case reported by Hof et al. (1993a). Dots emphasize the profound atrophy in the occipitoparietal region in this case. (B) Histology from another case of posterior cortical atrophy (ADB) in tau-ir and Nissl-stained tissues. Tau-ir neuropil threads and neuritic plaques were more dense in areas 29 and 30 of posterior cingulate cortex than in medial parietal area 7m. The density of NFT in layer Va was approximately equal in areas 23a and 7m. Neuron degeneration, however, was significantly different in these areas, with area 23a having severe degeneration in layer Va (asterisk) that did not occur in area 7m (Ctrl = age-matched control case). Thus, the density of tau-ir NFT do not accurately reflect total neurodegeneration, and the AD pathogenic process could have started in PCC rather than parietal cortex because of the longer standing (more severe) neurodegeneration.
In summary, these atypical AD cases serve as excellent examples of clinicopathological correlations between SP and NFT distributions and clinical symptoms that are likely to involve disruption of subsets of corticocorical connections. They are consistent with the observation of Victoroff et al. (1944) that posterior cortical atrophy is clinically homogeneous but displays neuropathologic heterogeneity. The disturbances in motion perception and target tracing frequently observed in AD (Neary and Snowden, 1987; Sadun et al., 1987; Fletcher and Sharpe, 1988; Katz and Rimmer, 1989; Kiyosawa et al., 1989; Butter et al., 1996) are more severe in posterior cortical atrophy and may be related to the very high densities of lesions in areas MT, 7b/7m, and 23. Although additional studies of such cases using functional imaging with provide key information that is missing between the neurologic symptoms and the affected critical circuits, current observations suggest close relationships between functional subsystems, connectivity patterns, and pathogenic mechanisms. The association of impaired cortical subsystems, gradients in NFT and Ab peptides, and clinical outcomes suggests that posterior cortical atrophy may be a subtype of AD.
In summary, these atypical AD cases serve as excellent examples of clinicopathological correlations between SP and NFT distributions and clinical symptoms that are likely to involve disruption of subsets of corticocorical connections. They are consistent with the observation of Victoroff et al. (1944) that posterior cortical atrophy is clinically homogeneous but displays neuropathologic heterogeneity. The disturbances in motion perception and target tracing frequently observed in AD (Neary and Snowden, 1987; Sadun et al., 1987; Fletcher and Sharpe, 1988; Katz and Rimmer, 1989; Kiyosawa et al., 1989; Butter et al., 1996) are more severe in posterior cortical atrophy and may be related to the very high densities of lesions in areas MT, 7b/7m, and 23. Although additional studies of such cases using functional imaging with provide key information that is missing between the neurologic symptoms and the affected critical circuits, current observations suggest close relationships between functional subsystems, connectivity patterns, and pathogenic mechanisms. The association of impaired cortical subsystems, gradients in NFT and Ab peptides, and clinical outcomes suggests that posterior cortical atrophy may be a subtype of AD.
Layer-Selective Neurodegeneration in Posterior Cingulate Cortex
Cases of AD with lobar degeneration suggest unique mechanisms of neurodegeneration to the extent that specific subsystems are involved and that each subsystem requires unique mechanisms to produce isolated patterns of cell death. Another conclusion from this survey is that PCC is profoundly impacted in the multifocal disease process(es). Neurofibrillary degeneration is often greater in PCC than it is in lateral neocortical areas, i.e., those areas that are usually invoked as a rationale for impaired functions. Glucose hypometabloism is very early AD is greater in PCC than in lateral neocortex and the parahippocampal gyrus (Minoshima et al., 1997), and it may be related to disorientation in time and place (Hirono et al., 1988). Thus, an early and profound impact of AD in PCC is suggested by both cortical atrophies and glucose metabolism. Furthermore, PCC is engaged in learning and memory (Valenstein et al., 1987) and in visuospatial functions (Murray et al., 1989; Olson et al., 1993). Hence, functional impairments often attributed to parietal and medial temporal cortices may commonly result form very early disruption of PCC.
Another approach to neuropathological heterogeneity, multifocal degeneration, and possible subtypes within AD is assessment of a consistently sampled cortical area in a large group of cases. Such an analysis does not begin with parahippocampal areas because this region appears to undergo a stereotyped pattern of neurodegeneration that is closely linked to NFT formation. In contrast, although the posterior cingulate region has been implicated in may studies of AD, the range of variability in each data set, including neurochemical, ligand binding, NFT, and neuron density, is extraordinary. Does such variability simply reflect a common theme, such as large neuron degeneration, or does it represent qualitatively different processes, such as large, medium, and/or small neuron degeneration early in the disease according to unique subtype mechanisms? There are five reasons for assessing PCC in this context and for approaching AD from a cingulocentric perspective.
First, PCC has a well-documented involvement in AD, including neurodegeneration (Brun and Englund, 1981; Mountjoy et al., 1983; Vogt et al., 1990), NFT and SP formation (Mountjoy et al., 1983; Vogt et al., 1990; Braak and Braak, 1993), and has significant reductions in choline acetyltransferase activity (Rossor et al., 1982; Procter et al., 1988).
Second, postmortem samples can be more consistently sampled from PCC than from any other neocortical region, because the spelenium of the corpus callosum provides a uniform landmark, standardizing tissue collection form multiple institutions.
Third, there is a full range of cytoarchitectural differentiation form periallocortical areas 26-30 to neocortical area 23. These areas are differentially impacted in AD and provide insight into disease progression (Vogt et al., 1990; Braak and Braak, 1993).
Fourth, there are strong connections between PCC and mid-prefrontal, posterior parietal, superior temporal, and parahippocampal cortices. This ensures that cingulate cortex is linked directly to those regions that have most frequently been implicated in the disease.
Fifth, PET studies show that PCC may be one of the earliest cortical regions to be involved in AD, including individuals with the earliest memory-loss symptoms (Minoshima et al., 1997).
Neurodegeneration is a better measure of cognitive decline than SP or NFT densities (Neary et al., 1986; Mann et al., 1988; Bennett et al., 1993), and the highest degree of variability in neuron degeneration is in PCC (0-80% losses; Brun and Englund, 1981). Although this latter study interpreted changes in terms of a graded disease progression in a single-etiology model, a subsequent study of PCC failed to support this view (Vogt et al., 1990). Neurodegeneration does not follow a single bilaminar pattern, and NFT and SP are a poor reflection of total neurodegeneration. Rather, there are qualitatively different laminar patterns in neurodegeneration in PCC and they may represent subtypes.
A consideration of the subtype hypothesis employed principal components analysis (PCA) and pattern-recognition methods to assess the extent to which laminar patterns of neurodegeneration represent statistically unique subgroups (Vogt et al., 1998).
Another approach to neuropathological heterogeneity, multifocal degeneration, and possible subtypes within AD is assessment of a consistently sampled cortical area in a large group of cases. Such an analysis does not begin with parahippocampal areas because this region appears to undergo a stereotyped pattern of neurodegeneration that is closely linked to NFT formation. In contrast, although the posterior cingulate region has been implicated in may studies of AD, the range of variability in each data set, including neurochemical, ligand binding, NFT, and neuron density, is extraordinary. Does such variability simply reflect a common theme, such as large neuron degeneration, or does it represent qualitatively different processes, such as large, medium, and/or small neuron degeneration early in the disease according to unique subtype mechanisms? There are five reasons for assessing PCC in this context and for approaching AD from a cingulocentric perspective.
First, PCC has a well-documented involvement in AD, including neurodegeneration (Brun and Englund, 1981; Mountjoy et al., 1983; Vogt et al., 1990), NFT and SP formation (Mountjoy et al., 1983; Vogt et al., 1990; Braak and Braak, 1993), and has significant reductions in choline acetyltransferase activity (Rossor et al., 1982; Procter et al., 1988).
Second, postmortem samples can be more consistently sampled from PCC than from any other neocortical region, because the spelenium of the corpus callosum provides a uniform landmark, standardizing tissue collection form multiple institutions.
Third, there is a full range of cytoarchitectural differentiation form periallocortical areas 26-30 to neocortical area 23. These areas are differentially impacted in AD and provide insight into disease progression (Vogt et al., 1990; Braak and Braak, 1993).
Fourth, there are strong connections between PCC and mid-prefrontal, posterior parietal, superior temporal, and parahippocampal cortices. This ensures that cingulate cortex is linked directly to those regions that have most frequently been implicated in the disease.
Fifth, PET studies show that PCC may be one of the earliest cortical regions to be involved in AD, including individuals with the earliest memory-loss symptoms (Minoshima et al., 1997).
Neurodegeneration is a better measure of cognitive decline than SP or NFT densities (Neary et al., 1986; Mann et al., 1988; Bennett et al., 1993), and the highest degree of variability in neuron degeneration is in PCC (0-80% losses; Brun and Englund, 1981). Although this latter study interpreted changes in terms of a graded disease progression in a single-etiology model, a subsequent study of PCC failed to support this view (Vogt et al., 1990). Neurodegeneration does not follow a single bilaminar pattern, and NFT and SP are a poor reflection of total neurodegeneration. Rather, there are qualitatively different laminar patterns in neurodegeneration in PCC and they may represent subtypes.
A consideration of the subtype hypothesis employed principal components analysis (PCA) and pattern-recognition methods to assess the extent to which laminar patterns of neurodegeneration represent statistically unique subgroups (Vogt et al., 1998).

Five subgroups were identified based on neurons in each layer in 160-mm-wide traverses of area 23a; neurons in each layer were not corrected for atrophy for reasons discussed in the article cited. Because each subgroup represented a different pattern of neuron loss that may require different pathogenic yet unknown events, they were referred to as subtypes (ST) within AD as follows: STO, no neurodegeneration; STIIIab, losses mainly in layer IIIab; STIV-V, losses mainly in layers IV-V; STII-V, losses of about 60% in most layers; STSevere, losses of more than 80% in most layers with almost no evidence of laminar architecture. The latter two subtypes are not different only in terms of total neurodegeneration, because they differ in age of disease onset (STSevere = 63 ± 3.4; STII-V = 74 ± 1.4; mean ± SEM), proportion of ApoE e4 homozygotes (STSevere = 42%; STII-V = 20%), SP (STSevere = 40 ± 3.9/1.9mm2; STII-V = 27 ± 1.8), and NFT (STSevere = 47 ± 9/mm2; STII-V = 20 ± 4.5).
The PCA was used to determine a principal components score, or eigenvector, for cases using neuron counts in layers IIIab, IIIc, IV, and Va. A three-dimensional plot, or eigenvector projection, of these values in Figure 6 shows that most AD cases segregate from the controls (Fig. 6A) and that, following removal of the controls to free statistical space in the eigenvector projection, 5 AD subgroups are formed (Fig 6B) as predicted form previous studies. Neurons for layers II, Vb, and VI have been explored to assess their contributions to improving subgrouping of cases (Fig. 6C), however, they did not improve the analysis, and layer VI has a wide range of neuron densities because it is near the apex of the ventral bank of the cingulate gyrus (i.e., densities are overwhelmingly affected by curvature). The individuals in Figure 6 are coded according to our previous a priori assessment of neurodegeneration by determining layers with greatest proportionate loss of neurons in comparison to controls. The fact that there is no overlap among the members of each subgroup suggests that the multiplayer model of neurons defines three subgroups. To the extent that each laminar pattern reflects unique mechanisms of cell death, this model defines subtypes within AD.
The PCA was used to determine a principal components score, or eigenvector, for cases using neuron counts in layers IIIab, IIIc, IV, and Va. A three-dimensional plot, or eigenvector projection, of these values in Figure 6 shows that most AD cases segregate from the controls (Fig. 6A) and that, following removal of the controls to free statistical space in the eigenvector projection, 5 AD subgroups are formed (Fig 6B) as predicted form previous studies. Neurons for layers II, Vb, and VI have been explored to assess their contributions to improving subgrouping of cases (Fig. 6C), however, they did not improve the analysis, and layer VI has a wide range of neuron densities because it is near the apex of the ventral bank of the cingulate gyrus (i.e., densities are overwhelmingly affected by curvature). The individuals in Figure 6 are coded according to our previous a priori assessment of neurodegeneration by determining layers with greatest proportionate loss of neurons in comparison to controls. The fact that there is no overlap among the members of each subgroup suggests that the multiplayer model of neurons defines three subgroups. To the extent that each laminar pattern reflects unique mechanisms of cell death, this model defines subtypes within AD.
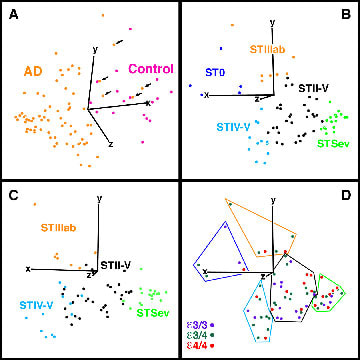
Figure 6. Eigenvector projections of principal components scores of neurons in layers IIIab, IIIc, IV, and Va. Graphs were rotated to demonstrate optimally the statistical subgroups. Click here to view the 3-D rotation of B.
(A) All AD (orange) and control (purple) cases; the 5 AD cases that overlap with the controls (arrows) had neuron counts like controls. (B) Each subtype is color coded according to layer-by-layer analysis of neurons which produced 5 subgroups: ST0 (purple), STIIIab (orange), STIV-V (blue), STII-V (black), STSevere (STSev, green). (C) Layer II was added to determine if it provided subgroup segregation. Since there was no improvement and the STIV-V/STII-V border was blurred, the multivariate model need not include neurons in this layer. (D) A test that ApoE genotype has a specific role in neuron losses is adding an ApoE risk score for each case and color code the data according to genotype as shown. There was no evidcnece for subgrouping by genotye except in STSevere.
Subtypes defined by neurons in layers III-Va are independent entities and do not simply merge with long durations of the disease. If this did occur, of course, there would be substantial overlap of cases, particularly at the perimeter of each subgroup in the eigenvector projections, but this is not so. Direct demonstration of subtype independence is provided in Figure 7, which shows perikaryal drawings for individual cases with different disease durations and from each subtype. Differences among the three subtypes can be visualized in this figure, and it can be seen the loss of neurons in any layer is essentially the same for short- and long-duration cases, i.e., 2-3 and 10 years, respectively. Finally, the range of the disease durations is similar (STIIIab, 3-15; STIV-V, 2-10; STII-V, 1-14 years), the age at disease onset is not significantly different (STIIIab, 69 ± 4; STIV-V, 75 ± 2; STII-V, 74 ± 2 years), and SP densities, verifying their inclusion under the diagnostic AD umbrella (STIIIab, 32 ± 2; STIV-V, 22 ± 5; STII-V, 27 ± 2).
(A) All AD (orange) and control (purple) cases; the 5 AD cases that overlap with the controls (arrows) had neuron counts like controls. (B) Each subtype is color coded according to layer-by-layer analysis of neurons which produced 5 subgroups: ST0 (purple), STIIIab (orange), STIV-V (blue), STII-V (black), STSevere (STSev, green). (C) Layer II was added to determine if it provided subgroup segregation. Since there was no improvement and the STIV-V/STII-V border was blurred, the multivariate model need not include neurons in this layer. (D) A test that ApoE genotype has a specific role in neuron losses is adding an ApoE risk score for each case and color code the data according to genotype as shown. There was no evidcnece for subgrouping by genotye except in STSevere.
Subtypes defined by neurons in layers III-Va are independent entities and do not simply merge with long durations of the disease. If this did occur, of course, there would be substantial overlap of cases, particularly at the perimeter of each subgroup in the eigenvector projections, but this is not so. Direct demonstration of subtype independence is provided in Figure 7, which shows perikaryal drawings for individual cases with different disease durations and from each subtype. Differences among the three subtypes can be visualized in this figure, and it can be seen the loss of neurons in any layer is essentially the same for short- and long-duration cases, i.e., 2-3 and 10 years, respectively. Finally, the range of the disease durations is similar (STIIIab, 3-15; STIV-V, 2-10; STII-V, 1-14 years), the age at disease onset is not significantly different (STIIIab, 69 ± 4; STIV-V, 75 ± 2; STII-V, 74 ± 2 years), and SP densities, verifying their inclusion under the diagnostic AD umbrella (STIIIab, 32 ± 2; STIV-V, 22 ± 5; STII-V, 27 ± 2).
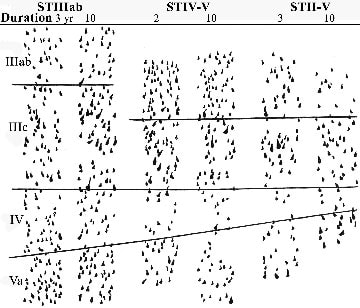
Figure 7. Neurodegeneration in area 23a of AD is not uniform, although it is relatively stable over the course of the clinical expression of the disease shown by perikaryal strips from six cases. In STIIIab the greatest neuron losses are in layer IIIab for 3- and 10-year durations. In STIV-V, greatest degenerations are in the deep cortical layers, whereas in STII-V, degeneration is significant throughout the cortex, with no laminar preference. There is no means of accounting for these patterns and progressions with a single-disease state that has a single progression.
Because PCC is significantly impacted in AD, it must be considered to what extent these impairments are reflected throughout neocortex. Perikaryal strips were analyzed through the right hemisphere of case WJ (presented in detail earlier). This hemisphere had no focal atrophy, Figure 8 shows samples from a number of neocortical areas. Although neuron losses occur in each layer, they are most profound in layers IV and Va, and there is a more limited loss in layer IIIab. This same distribution of neurodegeneration occurs throughout neocortex in this hemisphere, verifying that disruptions in PCC can reflect alterations throughout the neocortex.
Because PCC is significantly impacted in AD, it must be considered to what extent these impairments are reflected throughout neocortex. Perikaryal strips were analyzed through the right hemisphere of case WJ (presented in detail earlier). This hemisphere had no focal atrophy, Figure 8 shows samples from a number of neocortical areas. Although neuron losses occur in each layer, they are most profound in layers IV and Va, and there is a more limited loss in layer IIIab. This same distribution of neurodegeneration occurs throughout neocortex in this hemisphere, verifying that disruptions in PCC can reflect alterations throughout the neocortex.
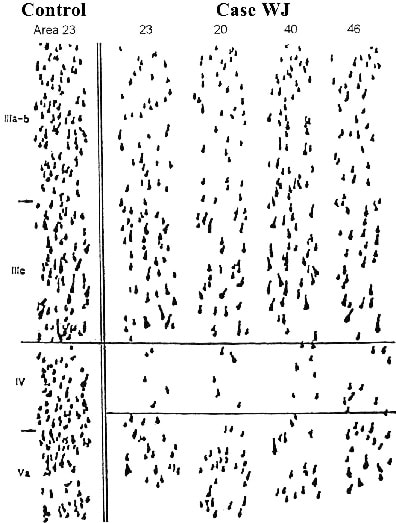
Figure 8. Neurodegeneration in area 23a of AD is not uniform, although it is relatively stable over the course of the clinical expression of the disease shown by perikaryal strips from six cases. In STIIIab the greatest neurodegeneration is in layer IIIab for 3- and 10-year durations. In STIV-V, greatest degenerations is in the deep cortical layers, whereas in STII-V, degeneration is significant throughout the cortex, with no laminar preference. Thus area 23a shows loss of neurons mainly in layers IV and V, and a more restricted loss in layer IIIab. This same pattern is reflected throughout lateral neocortical areas.
There is no means of accounting for these patterns and progressions with a single-disease state that has a single progression.
Because homozygous expression of the ApoE e4 allele is associated with elevated densities of SP (Schmechel et al., 1993; Sparks et al., 1996) and severe neurodegeneration in PCC (Vogt et al., 1998), it might be considered whether a direct link can be established between ApoE genotype and neurodegeneration. Subtypes based on laminar patterns of PCC neurodegeneration are ideal for direct tests of the relevance of the e4 allele to neocortical neuron losses. First, although there are significant differences in the laminar distribution of neurodegeneration in the 5 subtypes discussed above, the proportion of cases homozygous for the e4 allele is about the same: STIIIab, 29%; STIV-V, 15%; STII-V, 20%. Second, there were enough e3 (n = 9) and e4 (n = 6) homozygotes in STII-v to compare total neurons in layers IIIab-Va statistically. The differences between these two groups (e4/4 = 97 ± 9; e3/3 = 117 ± 7) were not significant. Third, coding the cases in these 5 groups according to ApoE genotype (3/3, 3/4, 4/4) and visual inspection of the eigenvector projection for neurons in layers III-Va for each shows that there is no subgrouping of cases according to ApoE genotype. Finally, an ApoE risk factor e2/ e3 = 2, e3/ e3 = 3, e2/ e4 = 4, e3/ e4 = 5, e4/ e4 = 6) in the neuron model does not identify new subgroups, nor does it alter subgroups defined with neurons alone (Fig. 6). Thus, ApoE genotype is not related to laminar specificities in neurodegeneration, although more e4 homozygotes occur in STSevere. Observations by Blacker et al. (1997) that the homozygous e4 risk is expressed primarily at ages 60-65 and lack of a role for heterozygous expression of the e4 allele suggest a restricted risk. Although there is an association between STSevere, with its early onset, high density of SP, and high proportion of e4 homozygotes, the ApoE e4 risk is not relevant to other subtypes with late-onset disease.
In conclusion, each subgroup, variant, or subtype of AD based on a unique laminar pattern of neuron losses progresses independently of the others. The different laminar patterns of neuron losses likely result from unique mechanisms for neurodegeneration. It is unlikely that any single protein, transmitter, or large-neuron model of AD etiopathology will account for the full breadth of AD heterogeneity; although the buildup of tau in particular groups of neurons appears to be critical for understanding differences in impairments of cognitive and affective domains of neuronal processing. For these reasons, complex statistical models are required to assess the clinical signs, genetics, and structural pathology expressed in this disease.
We hope you enjoyed this passage through a complex neurodegenerative disease in terms of its impact on the cingulate gyrus. Hopefully, you will not look at Alzheimer’s disease as a single entity and you will not expect a single treatment to be effective in all patients at all stages of the disease. Furthermore, you should be suspect of claims that any neuronal disease is a uniform entity and can be treated with a single drug therapy at all stages of disease progression.
There is no means of accounting for these patterns and progressions with a single-disease state that has a single progression.
Because homozygous expression of the ApoE e4 allele is associated with elevated densities of SP (Schmechel et al., 1993; Sparks et al., 1996) and severe neurodegeneration in PCC (Vogt et al., 1998), it might be considered whether a direct link can be established between ApoE genotype and neurodegeneration. Subtypes based on laminar patterns of PCC neurodegeneration are ideal for direct tests of the relevance of the e4 allele to neocortical neuron losses. First, although there are significant differences in the laminar distribution of neurodegeneration in the 5 subtypes discussed above, the proportion of cases homozygous for the e4 allele is about the same: STIIIab, 29%; STIV-V, 15%; STII-V, 20%. Second, there were enough e3 (n = 9) and e4 (n = 6) homozygotes in STII-v to compare total neurons in layers IIIab-Va statistically. The differences between these two groups (e4/4 = 97 ± 9; e3/3 = 117 ± 7) were not significant. Third, coding the cases in these 5 groups according to ApoE genotype (3/3, 3/4, 4/4) and visual inspection of the eigenvector projection for neurons in layers III-Va for each shows that there is no subgrouping of cases according to ApoE genotype. Finally, an ApoE risk factor e2/ e3 = 2, e3/ e3 = 3, e2/ e4 = 4, e3/ e4 = 5, e4/ e4 = 6) in the neuron model does not identify new subgroups, nor does it alter subgroups defined with neurons alone (Fig. 6). Thus, ApoE genotype is not related to laminar specificities in neurodegeneration, although more e4 homozygotes occur in STSevere. Observations by Blacker et al. (1997) that the homozygous e4 risk is expressed primarily at ages 60-65 and lack of a role for heterozygous expression of the e4 allele suggest a restricted risk. Although there is an association between STSevere, with its early onset, high density of SP, and high proportion of e4 homozygotes, the ApoE e4 risk is not relevant to other subtypes with late-onset disease.
In conclusion, each subgroup, variant, or subtype of AD based on a unique laminar pattern of neuron losses progresses independently of the others. The different laminar patterns of neuron losses likely result from unique mechanisms for neurodegeneration. It is unlikely that any single protein, transmitter, or large-neuron model of AD etiopathology will account for the full breadth of AD heterogeneity; although the buildup of tau in particular groups of neurons appears to be critical for understanding differences in impairments of cognitive and affective domains of neuronal processing. For these reasons, complex statistical models are required to assess the clinical signs, genetics, and structural pathology expressed in this disease.
We hope you enjoyed this passage through a complex neurodegenerative disease in terms of its impact on the cingulate gyrus. Hopefully, you will not look at Alzheimer’s disease as a single entity and you will not expect a single treatment to be effective in all patients at all stages of the disease. Furthermore, you should be suspect of claims that any neuronal disease is a uniform entity and can be treated with a single drug therapy at all stages of disease progression.